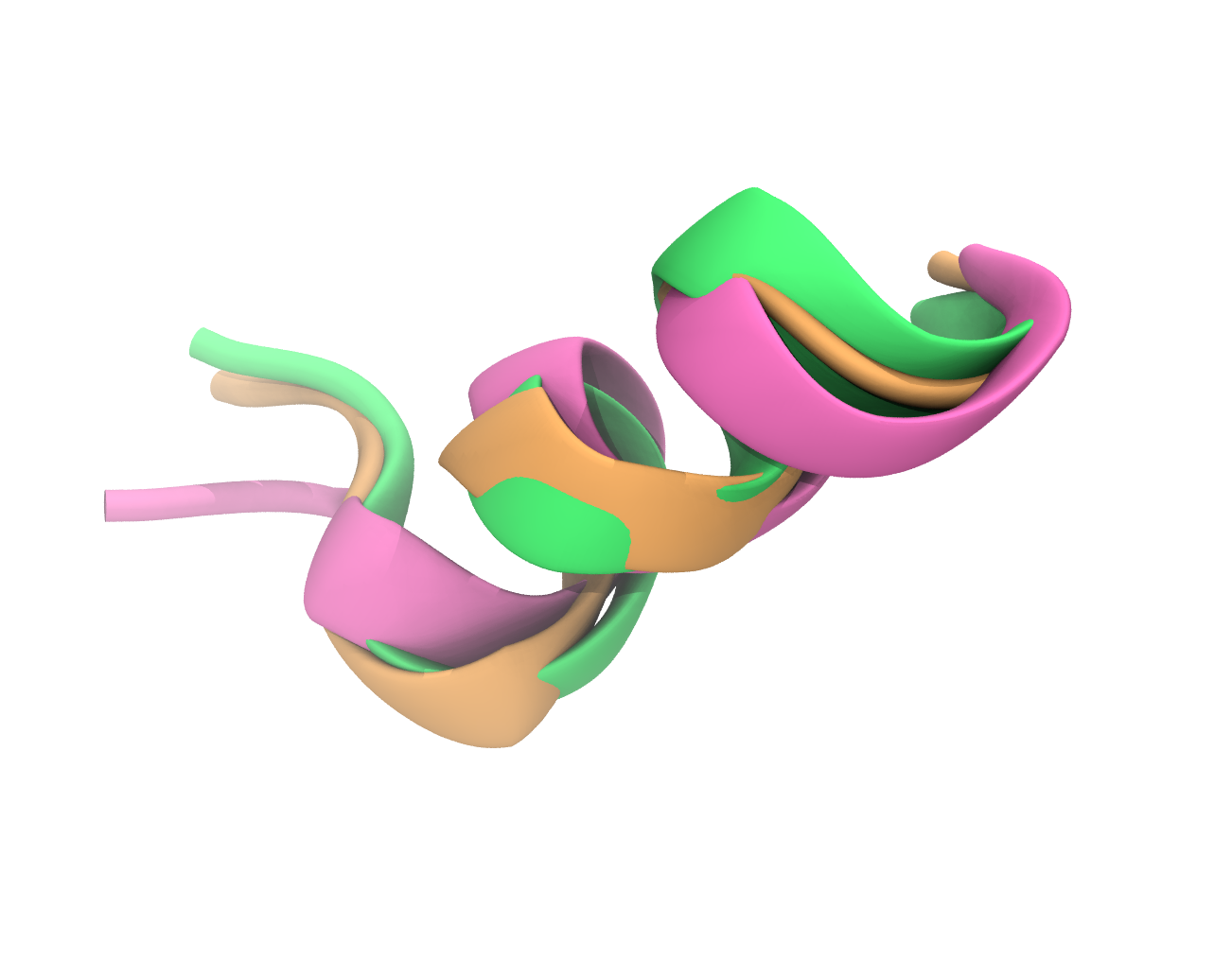Exercise: Molecular Dynamics with OpenMM – Solution¶
[3]:
from IPython.core.display import display, HTML
import matplotlib as mpl
import matplotlib.pyplot as plt
import simtk.openmm as mm # Main OpenMM functionality
import simtk.openmm.app as app # Application layer (handy interface)
import simtk.unit as unit # Unit/quantity handling
[4]:
# Jupyter notebook configuration
display(HTML("<style>.container { width:85% !important; }</style>"))
[5]:
# matplotlib configuration
mpl.rc_file("../../matplotlibrc")
Inspect the system¶
Go to the website of the RCSB protein data bank and download the solution NMR structure of a small peptide with the PDB-ID 6a5j as .pdb-textfile. Open the .pdb-file with the text editor of your choice and identify the section in which the molecular structure is described.
a) How many residues and atoms does the molecule have?
The peptide has 13 residues and 260 atoms.
[6]:
molecule = app.PDBFile("md_openmm/6a5j.pdb")
molecule.topology
[6]:
<Topology; 1 chains, 13 residues, 260 atoms, 259 bonds>
Parse the .pdb file with Python
[10]:
with open("md_openmm/6a5j.pdb") as pdbfile:
print("The structure contains:")
atom_count = 0 # How many atoms?
model_count = 0 # How many frames?
for line in pdbfile:
if line.startswith("SEQRES"):
print(f" {line.split()[3]} residues:")
print(f" {' '.join(line.split()[4:])}\n")
continue
if line.startswith("MODEL"):
model_count += 1
atom_count = 0
continue
if line.startswith("ATOM"):
atom_count += 1
continue
print(f" {atom_count} atoms\n")
print(f" {model_count} frames\n")
The structure contains:
13 residues:
ILE LYS LYS ILE LEU SER LYS ILE LYS LYS LEU LEU LYS
260 atoms
20 frames
b) Copy the atom definitions for the second amino acid residue and include it in your report.
[12]:
print_residue = 2
with open("md_openmm/6a5j.pdb") as pdbfile:
for line in pdbfile:
if not line.startswith("ATOM"):
continue
if int(line[22:26]) < print_residue:
continue
if int(line[22:26]) > print_residue:
break
print(line.strip())
ATOM 22 N LYS A 2 -7.295 3.130 -0.024 1.00 0.08 N
ATOM 23 CA LYS A 2 -6.396 2.084 -0.500 1.00 0.07 C
ATOM 24 C LYS A 2 -6.593 0.793 0.291 1.00 0.08 C
ATOM 25 O LYS A 2 -6.656 0.809 1.521 1.00 0.09 O
ATOM 26 CB LYS A 2 -4.934 2.531 -0.384 1.00 0.08 C
ATOM 27 CG LYS A 2 -4.546 3.653 -1.334 1.00 0.11 C
ATOM 28 CD LYS A 2 -3.114 4.126 -1.099 1.00 0.12 C
ATOM 29 CE LYS A 2 -3.033 5.105 0.061 1.00 0.62 C
ATOM 30 NZ LYS A 2 -3.301 4.453 1.372 1.00 1.15 N
ATOM 31 H LYS A 2 -6.928 3.882 0.486 1.00 0.19 H
ATOM 32 HA LYS A 2 -6.625 1.895 -1.537 1.00 0.09 H
ATOM 33 HB2 LYS A 2 -4.750 2.865 0.626 1.00 0.10 H
ATOM 34 HB3 LYS A 2 -4.298 1.682 -0.589 1.00 0.10 H
ATOM 35 HG2 LYS A 2 -4.633 3.296 -2.350 1.00 0.13 H
ATOM 36 HG3 LYS A 2 -5.218 4.485 -1.184 1.00 0.12 H
ATOM 37 HD2 LYS A 2 -2.484 3.270 -0.879 1.00 0.56 H
ATOM 38 HD3 LYS A 2 -2.756 4.613 -1.994 1.00 0.49 H
ATOM 39 HE2 LYS A 2 -2.043 5.536 0.080 1.00 1.15 H
ATOM 40 HE3 LYS A 2 -3.759 5.890 -0.097 1.00 1.06 H
ATOM 41 HZ1 LYS A 2 -2.685 3.624 1.493 1.00 1.67 H
ATOM 42 HZ2 LYS A 2 -4.292 4.147 1.422 1.00 1.71 H
ATOM 43 HZ3 LYS A 2 -3.118 5.123 2.147 1.00 1.58 H
Open the .pdb-file in VMD.
c) How many structures are contained in the file?
The .pdb-file contains 20 structures.
d) Set the background colour to white and remove the axis icon. Visualise the peptide structure in a representation that emphasises the secondary structure (e.g. NewCartoon, NewRibbons, …) and choose Secondary Structure as colouring method. Visualise the side chain of lysines in the structure in a representation that shows bonds and atoms (e.g. Licorice, CPK, …) and color them by chemical element. Render an image of the first structure frame and include it in your report.
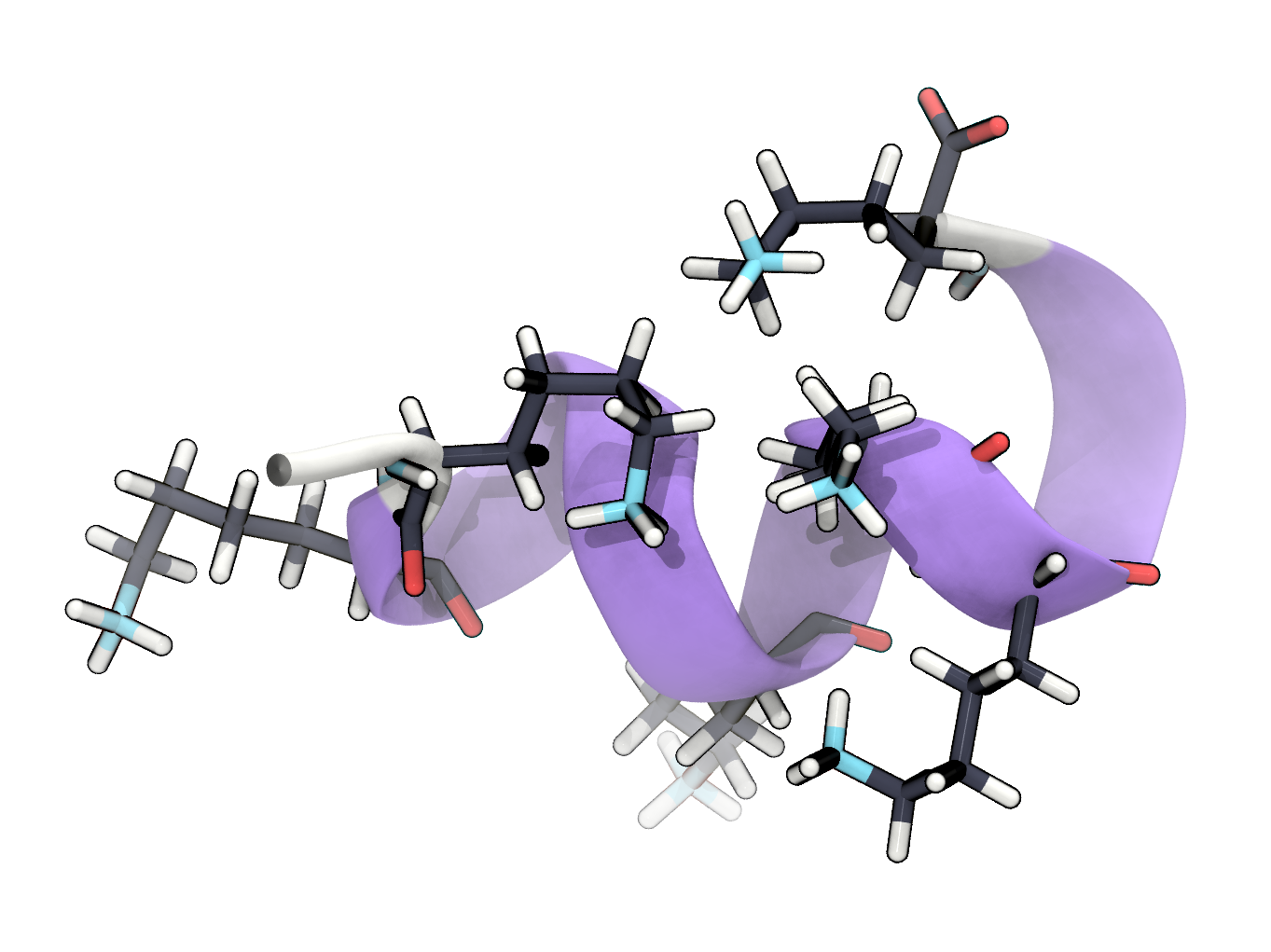
e) The structure file contains hydrogens. What is the total charge of the peptide?
Rational approach The peptide has 6 lysine residues in its sequence. All lysine side chains are protonated and the total charge of the molecule is +6.
Forcefield approach Get the total charge of the system from the partial charges of the atoms:
[7]:
forcefield = app.ForceField("amber14/protein.ff14SB.xml")
system = forcefield.createSystem(molecule.topology)
[8]:
for force in system.getForces():
print(type(force))
if isinstance(force, mm.openmm.NonbondedForce):
# For charges, we are only interested in the non-bonded forces
nonbonded = force
<class 'simtk.openmm.openmm.HarmonicBondForce'>
<class 'simtk.openmm.openmm.HarmonicAngleForce'>
<class 'simtk.openmm.openmm.PeriodicTorsionForce'>
<class 'simtk.openmm.openmm.NonbondedForce'>
<class 'simtk.openmm.openmm.CMMotionRemover'>
A non-bonded force entry of an atom looks like this:
[12]:
nonbonded.getParticleParameters(0)
[12]:
[Quantity(value=0.0311, unit=elementary charge),
Quantity(value=0.3249998523775958, unit=nanometer),
Quantity(value=0.7112800000000001, unit=kilojoule/mole)]
[9]:
force_description = zip(
["Partial charge", "Minimum energy distance", "Minimum energy"],
nonbonded.getParticleParameters(0)
)
for desc, comp in force_description:
print(f"{desc:<26} {comp}")
Partial charge 0.0311 e
Minimum energy distance 0.3249998523775958 nm
Minimum energy 0.7112800000000001 kJ/mol
[11]:
total_charge = 0.
for i in range(nonbonded.getNumParticles()):
nb_i = clePnonbonded.getPartiarameters(i)
total_charge += nb_i[0].value_in_unit(unit.elementary_charge)
total_charge
[11]:
5.999999999999995
[15]:
round(total_charge)
[15]:
6
Prepare the structure¶
Save the first structure included in the .pdb-file to a new separate .pdb file, e.g. conf.pdb. We want to use this frame as starting structure for our simulation. Use OpenMM to load the structure into memory.
[19]:
with open("md_openmm/6a5j.pdb") as pdbfile:
with open("md_openmm/conf.pdb", 'w') as newfile:
model_count = 0
for line in pdbfile:
if line.startswith("MODEL"):
model_count += 1
if model_count > 1:
break
continue
if not line.startswith("ATOM"):
continue
newfile.write(line)
[6]:
molecule = app.PDBFile("md_openmm/conf.pdb")
a) Investigate the topology attribute of the loaded molecule. How many covalent bonds are present?
[8]:
print(molecule.topology)
<Topology; 1 chains, 13 residues, 260 atoms, 259 bonds>
b) We want to simulate the molecule using the AMBER 14SB force field in TIP3P water. Create a forcefield object suitable for this purpose.
[52]:
forcefield = app.ForceField(
"amber14/protein.ff14SB.xml",
"amber14/tip3p.xml"
)
c) Our structure file does not include solvent and has a non-zero total charge. Put the molecule in a cubic water box with a minimum distance of 1 nm to the box boarders. Make sure that also counter ions are added to neutralise the system.
[5]:
model = app.Modeller(molecule.topology, molecule.positions)
model.addSolvent(
forcefield,
padding=1 * unit.nanometer,
neutralize=True
)
d) Save the modified structure (peptide plus solvent and ions) to a .pdb-file. Load the structure into VMD and visualise the peptide as before. Additionally show ions as van-der-Waals spheres and water molecules within a distance of 2 Å in the Licorice representation. Optional Add a transparent surface representation (e.g. QuickSurf) for the peptide.
[6]:
molecule.positions = model.getPositions()
molecule.topology = model.getTopology()
[7]:
with open("md_openmm/solv.pdb", "w") as file_:
molecule.writeFile(
molecule.topology, molecule.positions,
file=file_
)
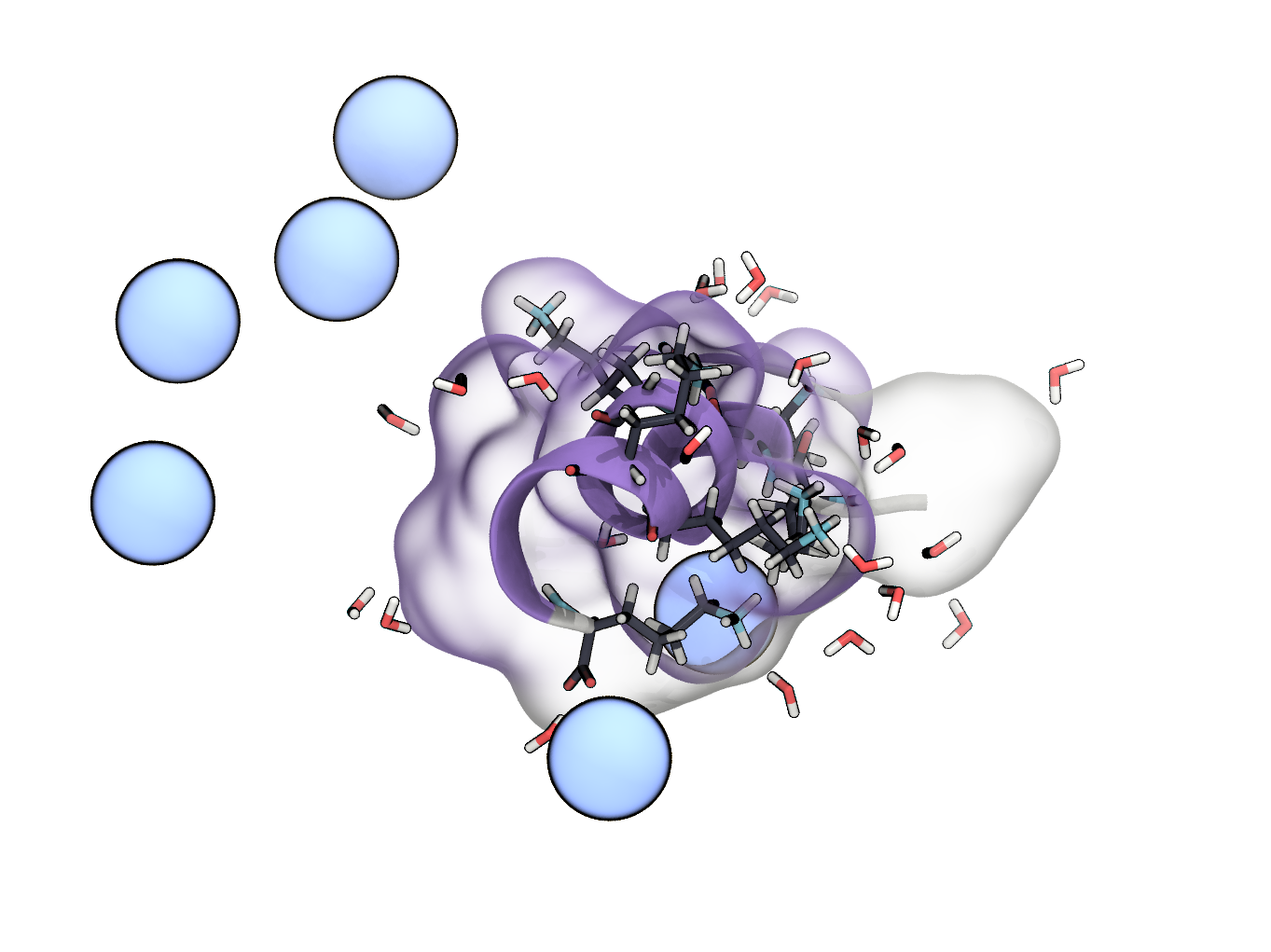
Simulation¶
Setup¶
a) Create a system object from the topology of the solvated peptide with the forcefield you setup earlier. Use H-bonds constraints, and PME for nonbonded interactions (cutoff 1 nm).
[50]:
molecule = app.PDBFile("md_openmm/solv.pdb")
[53]:
system = forcefield.createSystem(
molecule.topology,
nonbondedMethod=app.PME,
nonbondedCutoff=1 * unit.nanometer,
constraints=app.HBonds
)
b) We want to simulate in the NVT ensemble. Add an Andersen thermostat to the system to keep the temperature at 300 K (coupling rate 1/ps).
[54]:
system.addForce(
mm.AndersenThermostat(300 * unit.kelvin, 1 / unit.picosecond)
)
[54]:
5
c) We want to propagate the simulation using a Verlet integrator in timesteps of 2 fs (we need a contraint tolerance of 0.00001). Setup a simulation object with the solvated topology, the system and the integrator. Use
"CPU"as the simulation platform.
[55]:
integrator = mm.VerletIntegrator(2.0*unit.femtoseconds)
integrator.setConstraintTolerance(0.00001)
simulation = app.Simulation(
molecule.topology, # Topology
system, # System
integrator, # Integrator
mm.Platform.getPlatformByName('CPU') # Platform
)
How to figure out which platform to use?
[38]:
n_platforms = mm.Platform.getNumPlatforms() # How many platforms are supported?
n_platforms
[38]:
2
[39]:
for i in range(n_platforms):
print(mm.Platform.getPlatform(i).getName())
Reference
CPU
d) Add the current atomic positions of the solvated peptide to the context of the simulation.
[12]:
simulation.context.setPositions(molecule.positions)
Energy minimisation¶
a) Minimise the energy of the system. Save the new positions to a .pdb-file.
[13]:
simulation.minimizeEnergy()
[14]:
state = simulation.context.getState(getPositions=True)
molecule.positions = state.getPositions()
[15]:
with open("md_openmm/min.pdb", "w") as file_:
molecule.writeFile(
molecule.topology, molecule.positions,
file=file_
)
a) Open the structure in VMD and render an image the same way as in the last step in which the original and the minimised atoms are overlaid. Choose different colors for the two states. Can you see the difference?
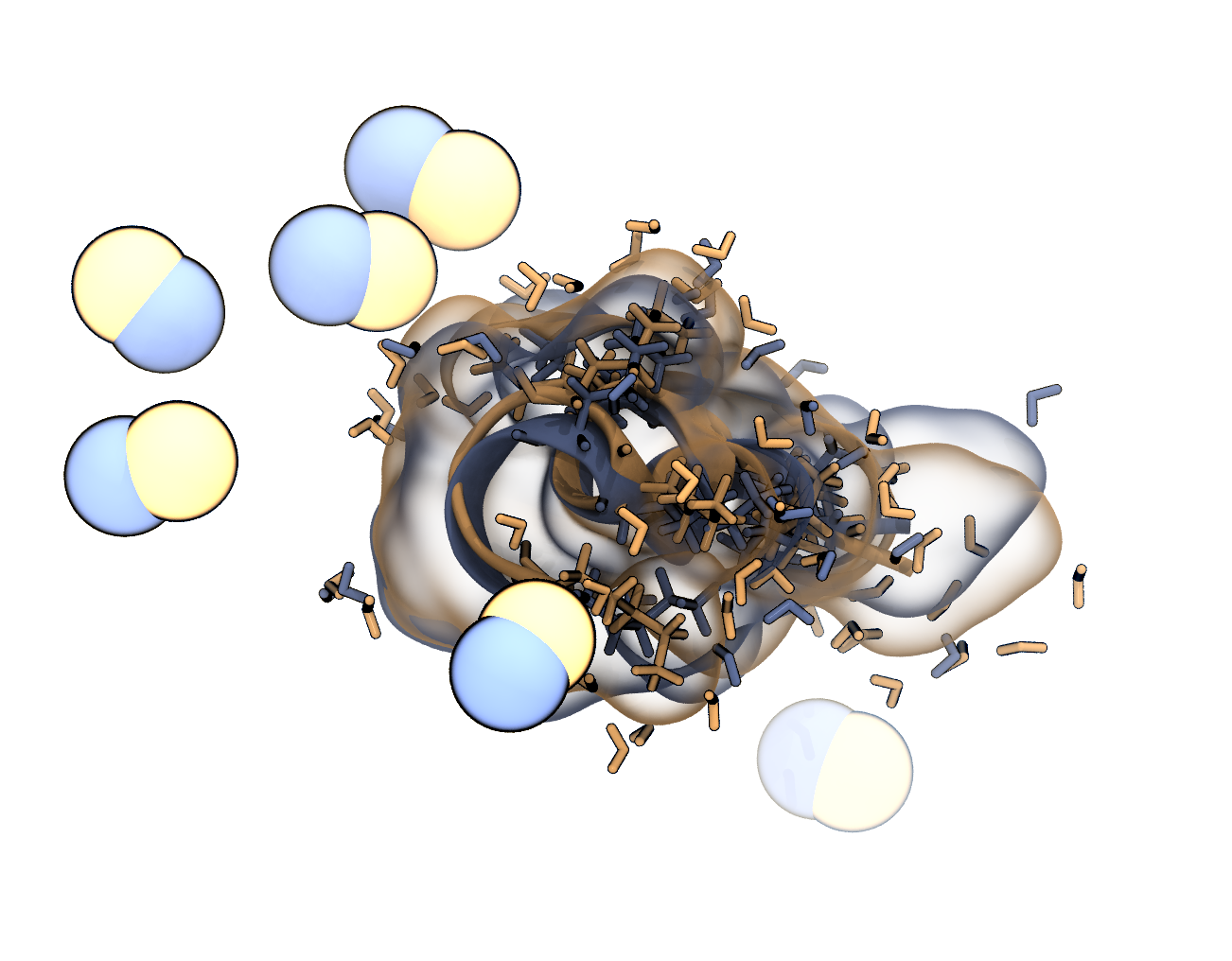
Equilibration¶
a) We now want to equilibrate our system in the NVT ensemble for 100 ps. Add a state reporter to the simulation object that reports the total and potential energy and the temperature at every picosecond and write it to a file (e.g.
equilibration.log).
[56]:
molecule = app.PDBFile("md_openmm/min.pdb")
simulation.context.setPositions(molecule.positions)
[17]:
run_length = 50000 # 50000 * 2 fs = 100 ps
simulation.reporters.append(
app.StateDataReporter(
"md_openmm/equilibration.log", 500, step=True,
potentialEnergy=True, totalEnergy=True,
temperature=True, progress=True,
remainingTime=True, speed=True,
totalSteps=run_length,
separator='\t')
)
b) Run the simulation. It should take about 10 minutes to complete on a processor with 4 cores. Reduce the number of steps, if it takes much longer on your hardware.
[18]:
simulation.step(run_length)
c) Plot the energies against the simulation time step.
[45]:
pot_e = []
tot_e = []
temperature = []
with open("md_openmm/equilibration.log") as file_:
for line in file_:
if line.startswith("#"):
continue
pot_e_, tot_e_, temperature_ = line.split()[2:5]
pot_e.append(float(pot_e_))
tot_e.append(float(tot_e_))
temperature.append(float(temperature_))
# Alternative solution:
# for l, v in zip((pot_e, tot_e, temperature), line.split()[2:5]):
# l.append(float(v))
t = range(1, 101)
[47]:
fig, ax = plt.subplots()
ax.plot(t, [x / 1000 for x in pot_e], label="potential")
ax.plot(t, [x / 1000 for x in tot_e], label="total")
ax.set(**{
"xlabel": "time / ps",
"xlim": (0, 100),
"ylabel": "energy / 10$^{3}$ kJ mol$^{-1}$"
})
ax.legend(
framealpha=1,
edgecolor="k",
fancybox=False
)
[47]:
<matplotlib.legend.Legend at 0x7f07884e3080>
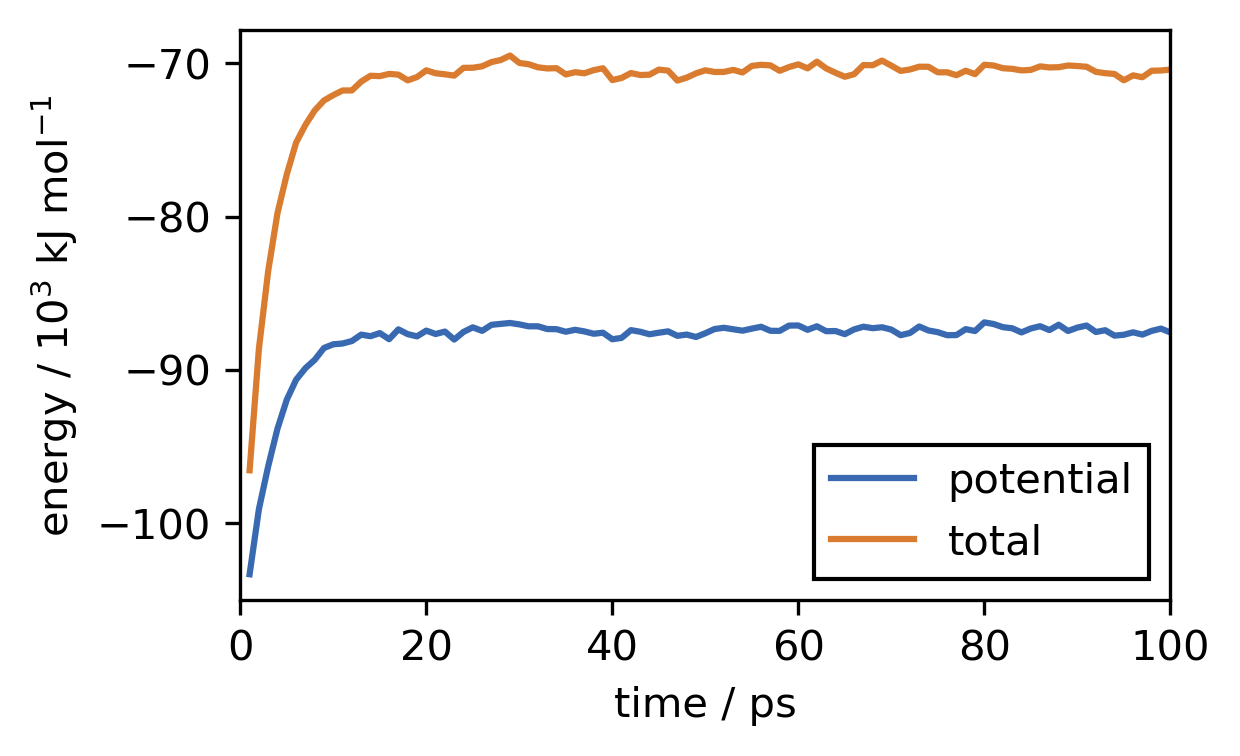
d) Plot the temperature against the simulation time step.
[48]:
fig, ax = plt.subplots()
ax.plot(t, temperature)
ax.set(**{
"xlabel": "time / ps",
"xlim": (0, 100),
"ylabel": "temperature / K"
})
[48]:
[Text(0, 0.5, 'temperature / K'), (0, 100), Text(0.5, 0, 'time / ps')]
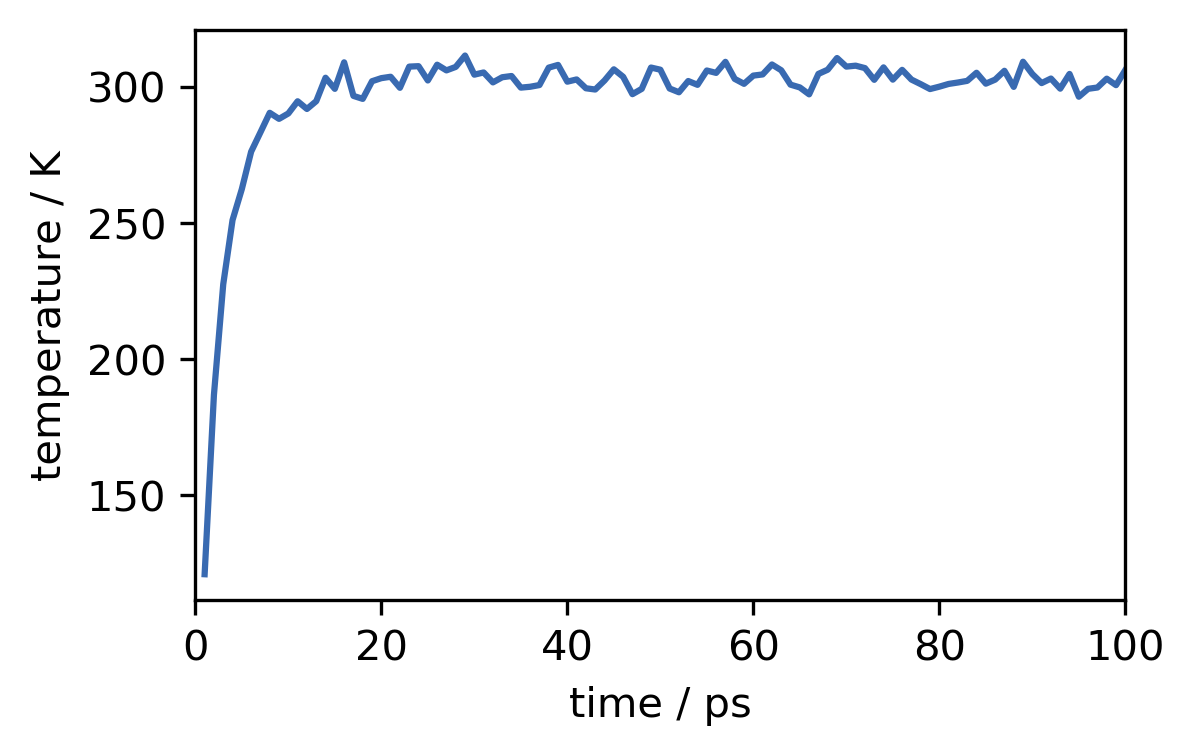
e) Save the current state of the simulation to an .xml-file.
[24]:
simulation.saveState("md_openmm/eq.xml")
Production¶
a) Load the state of the equilibrated system into the simulation object.
[57]:
simulation.loadState("md_openmm/eq.xml")
b) Reset the simulation reporters. Let’s now assume we want to simulate the system for 100 ns. Add a state reporter to the simulation object that reports the potential energy every 10 ps and writes it to a file (e.g.
simulation.log). Add a structure reporter that saves the current atom positions to a .pdb-file every 10 ps.
[26]:
simulation.reporters = []
run_length = 50000000 # 50000000 * 2 fs = 100 ns
simulation.reporters.append(
app.StateDataReporter(
"md_openmm/simulation.log", 5000, step=True,
potentialEnergy=True,
temperature=True, progress=True,
remainingTime=True, speed=True,
totalSteps=run_length,
separator='\t')
)
simulation.reporters.append(
app.PDBReporter('run.pdb', 5000)
)
c) The simulation can easily take several days to finish, depending on the used hardware, so you do not have to run it yourself completely. Just start the simulation to see if everything works and run it until the first structure has been saved by the structure reporter (after 10 ps).
[27]:
simulation.step(5100)
d) Open the saved .pdb-file in VMD. Render an image that shows only the peptide in a representation which emphasises the secondary structure and compares the original to the minimised and the current conformation. Align the structures (e.g. using the RMSD Trajectory Tool).